Help
Background
- MicroRNAs
- MicroRNA targets
- Artificial microRNAs
- Unique features of artificial microRNAs
- Hairpin RNAi versus microRNAs
AmiRNA Design Guide
- Quick Guide - amiRNA design overview
- Procedure - amiRNA design step by step
- Selection criteria
- Experimental procedure
- Target -Search-Tool
- Supplementary tools
Background
MicroRNAs
MicroRNAs (miRNAs) constitute a large group of endogenous single stranded small RNAs (19-24 nucleotides) that have negative regulatory function on gene expression. They are processed from longer non-coding RNAs with characteristic fold-back structures by double-strand specific RNases of the Dicer family. Upon processing, they are incorporated in the RNA induced silencing complex RISC by binding to its main component, an Argonaute protein. MiRNAs serve as the specificity components of RISC, since they base-pair to target nucleic acids, mostly mRNAs, in the cytoplasm. Subsequent regulatory events include target mRNA cleavage (often followed by degradation of cleavage products), which is mainly triggered by plant miRNAs, and translational inhibition, as mostly seen in animals. Target sites in plant miRNAs normally share high sequence complementarity to the respective miRNA and are often in coding regions, whereas in animals, target sites are often only partially complementary to their target sites and are mostly located in the 3'UTR of target genes. For further reading, we recommend Bartel (2004), Filipowicz (2005), Chen (2005), Jones-Rhoades et al. (2006) .
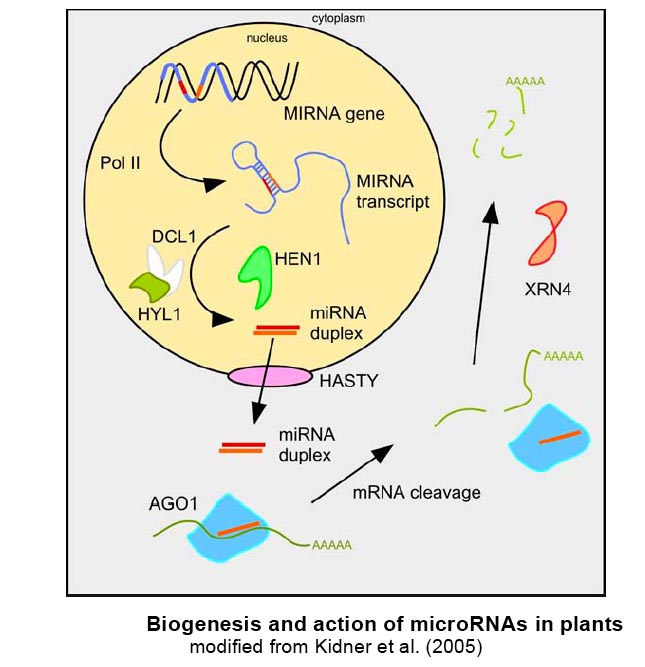
MicroRNA targets
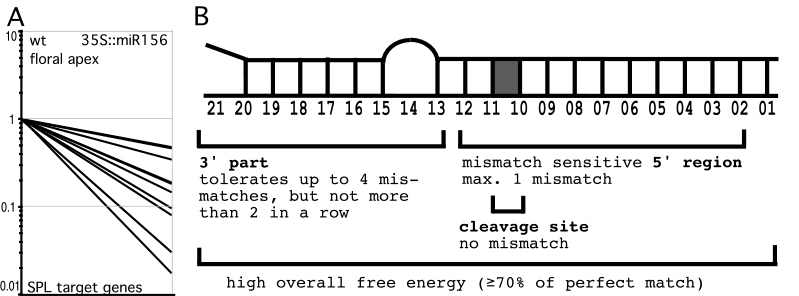
Plant miRNAs negatively regulate their targets (mainly protein-coding transcripts) by cleavage induced degradation. Effects of miRNA overexpression are thus often reflected in decreased mRNA levels of target genes (see Figure A).?In order to describe the full extend of plant miRNA targets, and to determine parameters of target selection, we have monitored genome-wide changes in transcript levels upon miRNA overexpression. Close inspection of genes with up to 5 mismatches to 5 different miRNAs revealed 2 groups: target genes, which are downregulated in miRNA overexpressers, and non-targets, which are not downregulated. Comparing alignments of all those genes to their respective miRNAs allowed us to determine parameters, which discriminate target genes from non-targets. They are summarized in Figure B and also hold true for most other known Arabidopsis miRNA-target pairs.
In animals, miRNAs normally pair to their targets with only limited complementarity, and recent publications support the view that base-pairing to the seed region between miRNA position 2 and 8 is sufficient for target recognition. Animal microRNAs therefore have many targets.
Reference for determinants of plant microRNA target selection:
Schwab et al. (2005)
References for animal microRNA target selection:
Brennecke et al. (2005),
Stark et al. (2005),
Lim et al. (2005),
Farh et al. (2005),
Lewis et al. (2005).
Artificial microRNAs
Artificial microRNAs (amiRNAs) are single-stranded 21mer RNAs, which are not normally found in plants and which are processed from endogenous miRNA precursors. Their sequences are designed according to the
determinants
of plant miRNA target selection, such that the 21mer specifically silences its intended target gene(s).?To compute optimal amiRNA sequences, we developed this WMD tool, which uses your favorite target genes as an input and generates all possible specific amiRNAs. AmiRNAs are required to resemble natural miRNAs by three criteria: they start with a U (found in most plant and animal miRNAs), they display 5' instability relative to their amiRNA* (the nucleotides pairing to themiRNA in the precursor), and their 10th nucleotide is either an A (most favorable according to
Reynolds et al. (2004)
for artificial siRNAs, and overrepresented in plant miRNAs (
Mallory et al. 2004
)) or an U.
For Arabidopsis, we use the MIR319a precursor to engineer amiRNAs by replacing the original miR319a (> the first 20 nts of the 21mer) with the artificial sequence (21mer), and the miR319a* with a sequence that pairs to the amiRNA with similar structural features than in the endogenous case. Arabidopsis precursors have been successfully used in tomato and tobacco
(Alvarez et al 2006)
, but we recommend to also test endogenous miRNA precursors from the species of interest for amiRNA engineering, preferentially those of which expression data is already available.
5' instability: Dicer generates small double stranded products with 2 nucleotide long 3' overhangs. One strand represents the miRNA, the other strand is called miRNA*. In the double strand, one end is normally thermodynamically less stable, since it is either mismatched or contains less GC (but more AU) pairs relative to the other end. The strand with lower thermodynamic stability at its 5' end (5' instability) is preferentially incorporated into RISC
Khvorova et al. 2003,
Schwarz et al. 2003
).
Unique features of artificial microRNAs
Since mainly one stable small RNA is generated from a miRNA precursor, silencing via this miRNA is highly sequence specific according to the determinants of plant miRNA target selection. This allows the design of amiRNAs, which silence either single or multiple related target genes. Related genes can also be located next to each other on the same chromosome (tandem repeats), and no other convenient tool exists for simultaneous silencing of these genes. As the recognition element within the target RNA is only very short, amiRNAs can also be utilized to silence specific splice forms of a gene of interest (see Design Guide below). Since miRNAs are single stranded, they mediate strand specific gene silencing. This allows the design of amiRNAs, which can specifically silence only antisense transcripts, which have remained largely unexplored to date. Similar to RNAi, amiRNAs also function in tissue-specific and inducible fashion, and they have only limited non-cell-autonomous effect. Most importantly, amiRNA induced mutants can be complemented by amiRNA resistant targets. Therefore, silent mutations are introduced in the region of amiRNA binding, which disrupt amiRNA mediated degradation. This technique has been first described by Palatnik et al. (2003) for natural miRNA targets.
Hairpin RNAi versus artificial microRNA
In Arabidopsis and other plants, directed gene silencing has often been approached by RNAi. Whereas initial experiments using overexpression of antisense transcripts (antisense to the gene which should be silenced) had only limited success, so-called hairpin constructs are now widely used and functional in many cases ( Wesley et al. 2001 For that purpose, 100-800bp fragments of the gene of interest are cloned in both orientations, separated by a loop-forming intron sequence, and expressed in planta. Primary transcripts form a fold-back structure, which is processed by Dicer enzymes into small RNAs. These are then perfectly complementary to the gene of interest and function by cleavage-mediated degradation of the target transcripts (for more information see http://www.pi.csiro.au/rnai/ ). Since transgenes are transcribed into long fold-back RNAs, many small RNAs are formed from one precursor. As the positions of Dicer cleavage are not known, the 5' ends of small RNAs (called siRNAs) are not known. In addition, it remains elusive, which parameters siRNAs use to select their target transcripts. Prediction of small RNA targets other then the perfectly complementary intended targets are therefore very hard to predict. Artificial microRNAs, in contrast, are produced from miRNA precursors, which are normally processed such that preferentially one single stable small RNA is generated. Since the determinants of plant miRNA target selection have been described, the complete spectrum of amiRNA targets is easily predictable. In addition, amiRNA sequences can be optimized for high efficiency, since they are always produced from the same locus in their precursors. Silencing of target genes by RNAi and amiRNAs is very similar and is based on cleavage mediated transcript degradation.
AmiRNA Design Guide
This paragraph describes how to use the WMD Design tool, and also how to proceed after amiRNAs have been designed. If you have never used the tool before, we strongly recommend reading this section!
Quick Guide - amiRNA design overview
The follwing list provides a swift overview of the steps involved in amiRNA design:- Please read the step by step guide to prevent frustration and to ensure optimal results.
- Blast your gene(s) of interest against the appropriate genome using the blast tool on this site. read more
- Identify the gene name used in the transcript or EST release up to the first blanc (i.e. AT1G01090 or TC203354 or Os12g29740.1 or 814761). read more
- Paste the target gene names into the "Target genes" field of the Designer page using the names identified in step 2 (do not use gene synonyms, abbreviations or any invented names for genes as the tool can not recognize them). read more
- Select a transcript library release.
- Fill out all required fields (email, description)
- If your gene could not be found in the blast search please submit your gene in regular fasta format. The gene name you use is not allow to be present in the transcript library!
- After submission you should receive an email with the results within a few hours (depending on server load). read more
- Click on each amiRNA candidate to see alignments of the amiRNA with all targets in the genome. read more
- Select the optimal amiRNA using these empirically found criteria.
- Read our Experimental Procedure Guide to learn more about oligo design, cloning of amiRNAs and plant transformation.
Procedure - amiRNA design step by step
-
Input sequences
WMD3 supports many plant species, however there are different input formats for annotated and non-annotated genes and additional settings to allow silencing of exogenous genes as well as spliceform specific silencing:
A) ANNOTATED GENES The first step to calculate AmiRNAs for one or more genes is to find out if the genes are annotated in the respective plant genome releases (e.g. At1g23450 in A. thaliana or Os01g24680 in O. sative) using WMD-Blast. The identified gene names have to be pasted into the "Target genes" field in exactly the same way (case sensitive) as found in the blast results. To silence several genes simultaneously, you also have to specify a minimal number of targets (2 or more) that should be targets of one amiRNA. WMD will automatically form subgroups of the target genes and deliver amiRNAs for those.
B) NON ANNOTATED GENES If the target genes could not be found in the respective transcript library using WMD-Blast, target genes have to be entered in a FASTA format (multiFASTA for multiple targets). The name used in the fasta header should not be equal to another gene from the respective transcript library. WMD3 will automatically check if your submission is correct fasta and if the used names are not redundant with other genes in the transcript library.
C) EXOGENOUS TARGETS For targets which are not from the selected genome releases, such as viral genes or other non-plant sequences like GUS or GFP, follow the exact same procedure as described in B)
D) SPLICEFORM SPECIFIC SILENCING Silencing of individual splice forms requires entry of the regions that are unique in the spliceform of interest in FASTA format. The header has to be a new name that is not occurring in the selected transcript library. In addition the real spliceform name has to be added as an acceptable off-target. While this might seem circuitous it is currently the only way to achieve spliceform specific silencing using WMD.
E) ACCEPTABLE OFF-TARGETS Genes and especially ESTs can often bee redundant which hampers the ability of WMD to produce specific amiRNAs. In other cases a highly similar gene could exists that is known to be non functional and which thus could be acceptable as off-target. The Acceptable off-target field therefore gives the possibility to define an indefinite number of transcripts that WMD does not count as off-targets when designing amiRNAs. The names of all redundant (highly similar) or non-functional transcripts have to be identified using WMD-Blast. -
Designer input form
-
Go to the "Designer" window.
-
Enter all target genes in the appropriate format (see Input sequences above) into the "Target genes" window. See the examples if you are not sure how this should look.
-
Select the genome release you are interested in.
-
Specify the minimal number of target genes (2 or more) when you want to silence multiple genes simultaneously. This helps the program to create subgroups when simultaneous silencing of all genes is not possible.
-
Specify any off-target as described in the "input sequences" section above in the format of the genome release that you selected (comma-separated).
-
You may enter comments in the description field. They are used as a title in your email so that you can recognize and distinguish your predictions.
-
Enter your email address. Your results will be emailed, so if you enter an invalid address they will never reach you.
-
Press the submit button. You will be directed to a page confirming the job. Calculations can be very fast or take several hours, depending on server occupancy. When we already have the results in our database, you will be promptly directed to those.
-
-
Output
Your email will contain a link to your result page. This page shows you a summary of your job at the top, followed by a list of amiRNA sequences (might be blank when you request did not yield any candidates, see instructions below). The list contains the amiRNA sequence (column 1, hyperlinked to an alignment to the target gene and other similar genes), the hybridization energy of the miRNA to a perfectly-matching complement (column 2), the identifier of the target gene (column 3), and the hybridization energy of the miRNA to the target site in the target gene (column 4). These amiRNAs are ranked by different criteria (i.e. amiRNA sequence composition (e.g.degree of 5' instability), mismatch positions when paired to intended target(s), hybridization energy when paired to intended target(s), number of other genes which have 5 or less mismatches to the amiRNA (not following the mismatch rules, preferably very few), hybridization energies of other genes with 5 or less mismatches to the amiRNA (not following mismatch rules, preferentially much higher), etc.). According to those criteria, we colored the presumably best ones in green, followed by intermediate ones in yellow and orange, and the remaining ones in red at the bottom of the list. Please note that red does not necessarily mean non-functional, but rather potentially less specific. We recommend to look at those amiRNAs from top to bottom.
In case you did not get any results back (result page only shows the job description):-
Your conditions might have been too stringent. If you wanted to silence multiple genes simultaneously, you might have to reduce the minimal number of included target genes and allow WMD to form smaller subgroups. If you gave an EST as a template target gene, it might have been too short to produce sufficient numbers of candidate sequences. You can try to assemble a pseudo EST as described in the input sequences section.
-
Your gene might be too similar to other genes to be silenced individually.
-
Your genes might be too different to silence them simultaneously.
You might want to download the Excel-File, which is available on the result page. It contains amiRNA candidates, which have not made it through the specificity filters. By copying the sequences into the TargetSearch tool, you can find out why they were rejected and change your input accordingly.
-
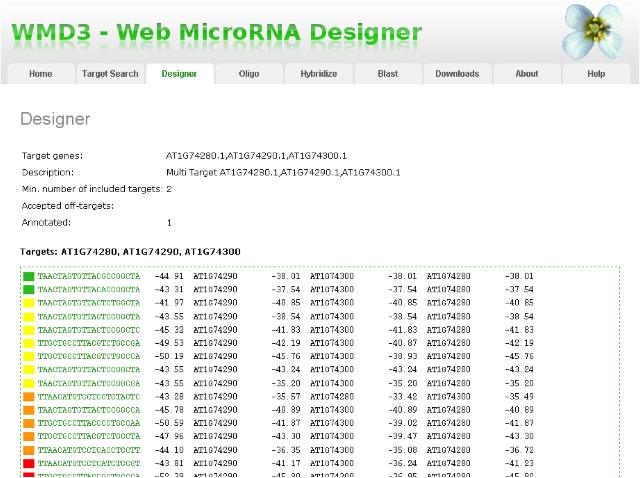
Result page of WMD3 amiRNA Designer: amiRNAs are sorted according to established quality criteria. AmiRNAs at the top of the list are expected to work better in most cases.
Selection criteria
There are still some criteria, which have to be considered when choosing the final amiRNA. Most of them have been implemented into the ranking process, and they should be considered here again, especially when multiple genes are targeted simultaneously.We prefer (not require):
-
No mismatch between positions 2 and 12 of the amiRNA for all targets.
Mismatches are not allowed for the target gene that is used as a template, but they might come up for additional intended targets since the target determinants allow for one mismatch. -
One (or two) mismatches at the amiRNA 3? end (pos.18-21).
There is no evidence for transitive formation of secondary siRNAs from amiRNA targets, but if there was, this mismatch should reduce the process. -
Similar mismatch pattern for all intended targets.
There is no evidence that the pattern of mismatches matters, but similar patterns definitely don't hurt. -
Absolute Hybridization energy between -35 and -38 kcal/mole.
These are the values observed for most endogenous miRNA targets. We don?t consider amiRNAs which pair to intended targets with energies higher than -30 kcal/mole. -
Target site position.
There is no evidence that the position of the target site in the target transcript has an effect on effectiveness, but target sites in most endogenous miRNA targets are found towards the 3' end of the coding regions. Examples in the 3'UTR are also not uncommon.
To facilitate the selection of amiRNAs, their sequences are hyperlinked to an alignment to all transcripts in the respective genome release that have up to 5 mismatches. They are sorted by hybridization energy between target gene and amiRNA, and your intended target gene(s) should appear on top of the list. The alignments show the target sequence (5'->3' orientation), the amiRNA as a reverse complement (amiRNA position 1 is on the right side), mismatch patterns (stars indicate matching bases), as well as the relative position of the target site in the transcript. Additional information is given by the hybridization energy between the respective target site and the amiRNA (should be as higher as possible in all genes which are not intended targets relative to the intended target), and the indication why a transcript has been rejected as a target gene (red bar).
Once selected, the amiRNA sequence needs to be engineered into an endogenous microRNA precursor for expression. We provide the "Oligo" tool, which designs primer for the Arabidopsis thaliana MIR319a (ath-miR319a) and Oryza sativa MIR528 (osa-miR528) precursors. You simply have to paste your amiRNA sequence and retrieve 4 oligo sequences (they are in 5'->3' orientation already), which are used for site-directed mutagenesis (see below). They retain structural features in the respective MIRNA precursors. For historical reasons, in the case of the ath-miR319a, we replace a 20mer, which was originally thought to represent ath-miR319a (we now know that it is rather preferentially 21nt long) by a 21nt sequence, thereby adding 1 pairing base at the bottom of the MIRNA stem. This does not interfere with miRNA production.
When you want to use other MIRNA precursors routinely or want to share your precursor with others, contact us (wmd@tuebingen.mpg.de) and we implement your precursor here as well! Generally, these oligos are 40nt long, comprising (5' to 3') two nucleotides matching the MIRNA precursor - 21nt replacing the miRNA (or miRNA*) - 17nt matching the MIRNA precursor. The overlap in the miRNA (and miRNA*) region is 25nt (2+21+2).
Experimental Procedure
-
Cloning
The first step is to engineer the amiRNA and amiRNA* sequences into an endogenous MIRNA precursor. We have generated a protocol for the Arabidopsis precursor MIR319a, which is available for download here and which can be transferred to any other starting precursor. It uses site-directed mutagenesis on a template plasmid containing the MIR319a precursor (pRS300, based on pBSK). This plasmid does not contain any promoter and can be requested from Prof. Weigel (weigel@tuebingen.mpg.de). A similar plasmid is available for rice and contains the osa-miR precursor.
After sequence confirmation, amiRNA precursor are placed behind a promoter of interest and transferred into a binary plasmid. Many restriction enzymes are available that flank the MIRNA319a precursor in the pRS300 plasmid (we often use EcoRI and BamHI sites), and it is also possible to engineer Gateway? sites when fusing the initial PCR products to directly recombine into suitable target plasmids. When you by accident cloned your amiRNA in reverse orientation, you have to go back and use different restriction enzymes. The secondary structure formed by MIRNA precursors only forms in the sense orientation.
We often use strong constitutive expression of amiRNAs (e.g. 35S promoter), but tissue-specific and inducible promoters have been tested successfully as well. It is important to mention that amiRNA-mediated gene silencing is quantitative at least in some respect, since stronger promoters can induce more dramatic effects than weaker ones. Therefore, we do not recommend using weak promoters when strongly expressed genes should be silenced efficiently. They might nevertheless become very useful when only partial silencing is intended.
We (and others) have used different sets of binary plasmids for Arabidopsis and have not had problems with any of them. Therefore, we recommend using a binary plasmid that is commonly used in the species of interest. -
Transformation
Binary plasmids containing amiRNA precursors should be transferred into plants using established transformation protocols. When target genes are also available as transgenes behind strong promoters, both can be simultaneously and transiently introduced into tobacco leaves to test for functionality. A detailed protocol is soon available here.
-
AmiRNA expressing plants
Primary transformants should be selected with appropriate selection markers. Due to different insertion sites, transgenes can be expressed at variable levels in different transgenic lines even when the same promoter was used. Therefore, primary transformants may vary in their phenotypes.
The lack of a phenotype can have different reasons. The mutant phenotype might not be detectable under the condition used to grow the plants (e.g. different condition needed, or mutant phenotype hidden due to genetic redundancy), or the downregulation of the target gene might not be sufficient to detect phenotypic changes.
Since miRNA mediated gene silencing effects are mostly evident on the transcript level, you can test the degree of target downregulation by RT-PCR, preferentially with oligos spanning the amiRNA target site. In few cases however, we have detected the expected phenotypic abnormalities without changes in target transcript expression, suggesting that amiRNAs might also function via translational inhibition on some target genes as do some endogenous miRNAs.
Weak promoters (weak in the region where the target gene is expressed) might not be sufficient to efficiently silence a target gene. We recommend to carefully select a promoter before generating transgenic plants.
A survey on amiRNA efficiency, which we have conducted in the spring of 2007, suggests that around 75% of amiRNAs designed with our current knowledge lead to silencing of target genes in Arabidopsis. We are working on different aspects to improve this success rate, but also recommend the generation of two amiRNAs per target gene(s), preferentially with target sites in different regions of the target transcripts. Two main reasons could account for insufficient gene silencing: (1) the inaccessibility of the amiRNA target site in the 3-dimensional context of the target transcript, and (2) difficulties in reducing the steady-state level of the target transcript(s) because of negative feedback regulation. Genes falling into the latter class might not be suitable for engineered post-transcriptional gene silencing. Studying the accessibility of target sites requires sophisticated predictions of RNA secondary structure, which is not trivial. However, attempts to study this issue are on the way and might improve amiRNA success rates in the near future. -
Propagation of amiRNA expressing plants
As far as we tested, amiRNA-mediated gene silencing was stable in following generations.
-
Phenotypic complementation of amiRNA expressing plants
Since target sites of amiRNA in target transcripts are small and distinct, it easily possible to engineer silent mutations (preferentially as many as possible) within the target site, such that the transcript is no longer susceptible to amiRNA mediated regulation. Re-introducing this transgene (endogenous or stronger promoter) should complement phenotypes, which are caused by amiRNA-mediated downregulation of this target gene. Silent mutations have been successfully used to release regulation by endogenous miRNAs, and they can provide evidence that phenotypes are caused by downregulation of the intended (and not any other) target gene when respective mutants are not available or not viable.
Target-Search-Tool Guide
TargetSearch is based on GenomeMapper, a short sequence alignment tool developed for the 1001 genomes project:
http://www.1001genomes.org
-
Search
Any DNA or RNA sequence can be used in target search to find all occurrencies of the sequence or the reverse complement of the sequence within a genome. You can click on example sequence to test TargetSearch.
-
Genome
You can choose between multiple plant EST or transcript releases. We do not provide searches against whole genomes because siRNAs target genes, however you can download the AmiRNA/WMD3 API and setup a local copy to enable this feature.
-
Mismatches
Target Search will return any genome sequence matching your input query with up to the number of mismatches specified here. If 'Allow gaps=Yes' is specified, the Mismatches field specifies the sum of mismatches and gaps allowed in the the alignment.
-
Apply microRNA Filter
Select 'Yes' to filter the results of the initial alignment using empirically determined features of miRNA target selection. The filter algorithm can further be customized using the fields 'Perfect-match-dG cutoff', 'Hybridization temperature' and 'Folding program'. Select 'No' to completely turn the miRNA filter of.
-
Perfect-match-dG cutoff
Set the minimum ratio of hybridization energy dG (miRNA/target) to the optimal hybridization energy in percent. Optimal hybridization energy is calculated using the miRNA sequence and its perfect reverse complement.
-
Hybridization temperature
Set the temperature for the calcualtion of RNA-RNA hybridization. Default is 23° C.
-
Folding program
WMD/AmiRNA currently supports two RNA folding programs both from the ViennaRNA package: RNAcofold and RNAup. The main difference of RNAup compared to RNAcofold is that RNAup takes into account the energy needed to unfold the target mRNA (a measure for approximation of the target site accessibility).
-
Direction
Alignments can be performed in forward or reverse complement orientation. For miRNA target search the default orientation is 'reverse'.
-
Allow gaps
Allow for gapped alignments. The total number of allowed gaps + mismatches is set using the 'mismatch' field.
-
Show flanking sequence
Return additional bases upstream and downstream of the target site.
-
Show only one isoform
If select only one isoform (spliceform) per gene is reported. This will not always be the first isoform but the first isoform which was found in the search process. If you would like to test if all isoforms contain the same (target) sequence set this option to 'No'.
-
Sort mode
The search results can be sorted by the evalue of the alignment or the hybridization energy (dG) of miRNA and target mRNA. Both sort modes can be selected in ascending and descending order.
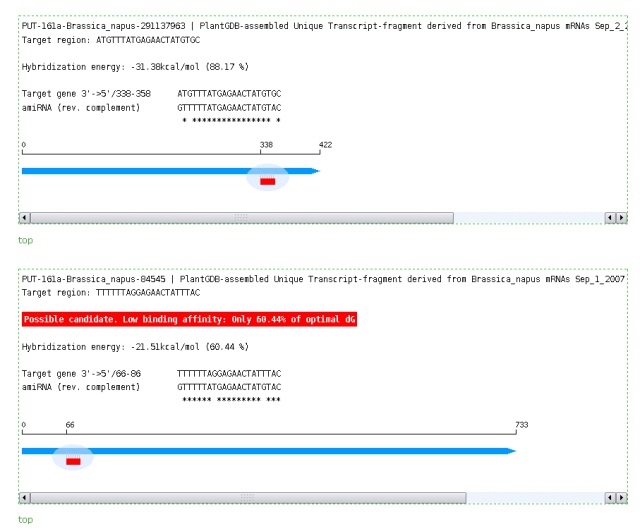
Output of WMD3 TargetSearch: Alignment of amiRNA against target mRNA,position of target site in the mRNA and free hybridization energy.
Supplementary Tools
-
Blast Tool
The Blast tool reads multi fasta sequences and blasts them against the selected transcript or EST release. This is useful to identify a gene of interest and the gene name within the current release before using it for amiRNA design. Blast can also identify redundant ESTs or potential off-targets in advance.

Output of Blast search of "some_gene" against EST database of Lycopersicon esculentum. The correct identifier that should be used for amiRNA design instead of "some_gene" is marked in blue.
-
Hybridization Tool
The hybridization tool is based on RNAcofold and RNAup from the Vienna RNA package. The tool calculates the free hybridization energy between two (independent) RNA sequences. The second input sequence can be reverse complemented automatically by selecting the checkbox "Rev-Comp Seq B". (Test this by pasting the same sequence into both sequence fields and select "Rev-Comp Seq B". This returns the "perfect" hybridization of the sequence with it's reverse complement). Additionally you can specify the temperature for which the hybridization should be calculated (i.e. 37° for human, 16° or 23° for A.thaliana).

Hybridization of an amiRNA to it's mRNA target site using RNAcofold.


